全球首例,!6個(gè)月嬰兒接受定制基因編輯治療 罕見病治療新突破

全球首例,!6個(gè)月嬰兒接受定制基因編輯治療 罕見病治療新突破。美國費(fèi)城兒童醫(yī)院和賓夕法尼亞大學(xué)醫(yī)學(xué)院的研究團(tuán)隊(duì)最近取得了一項(xiàng)具有里程碑意義的醫(yī)學(xué)突破——人類首次實(shí)現(xiàn)為單個(gè)病人定制基因編輯療法,,并成功治療了一名患有罕見致命遺傳疾病的嬰兒,。這項(xiàng)研究于2025年5月15日發(fā)表在《新英格蘭醫(yī)學(xué)雜志》上,,詳細(xì)介紹了這款定制的體內(nèi)堿基編輯療法的開發(fā)和治療過程。
這一成功案例可能為基因編輯技術(shù)開辟一條應(yīng)用于治療那些目前尚無醫(yī)療手段可用的罕見病患者的新途徑,。嬰兒KJ Muldoon在出生幾天后被診斷出患有嚴(yán)重的氨甲酰磷酸合成酶-1(CPS1)缺乏癥,。這是一種罕見且嚴(yán)重的隱性遺傳疾病,,在新生兒中的發(fā)病率為130萬分之一,是最嚴(yán)重的尿素循環(huán)障礙疾病之一,。人體在分解蛋白質(zhì)時(shí)會(huì)產(chǎn)生氨,,正常情況下會(huì)將氨轉(zhuǎn)化為尿素并排出體外。CPS1功能缺失會(huì)導(dǎo)致氨迅速在血液中積聚,,從而導(dǎo)致器官損傷,,尤其是大腦和肝臟,若沒有得到及時(shí)治療,,可能會(huì)迅速進(jìn)展為昏迷甚至死亡,。據(jù)統(tǒng)計(jì),CPS1缺乏癥患者在嬰兒早期的死亡率高達(dá)50%,。
CPS1基因很大,,超過了腺相關(guān)病毒(AAV)載體的遞送極限,此外,,嬰兒的肝細(xì)胞更替速度較快,,導(dǎo)致遞送到肝臟的載體會(huì)被快速稀釋。因此,,該疾病難以通過傳統(tǒng)的遞送正確版本的CPS1基因進(jìn)行治療,。目前,該疾病的治療手段有限,,包括透析,、氨清除劑、限制蛋白質(zhì)攝入以及晚期肝移植,,但這些方法難以阻止氨對(duì)大腦神經(jīng)系統(tǒng)的損傷,。
不久前,美國FDA批準(zhǔn)了基于CRISPR-Cas9的基因編輯療法Casgevy上市,,用于治療兩種相對(duì)常見的遺傳疾病——鐮狀細(xì)胞?。⊿CD)和β-地中海貧血(TDT)。堿基編輯是劉如謙教授基于CRISPR開發(fā)的新一代基因編輯技術(shù),,其不依賴DNA雙鏈斷裂,,能夠精準(zhǔn)修復(fù)人類基因組中的致病突變。

沐曦股份完成IPO輔導(dǎo) 國產(chǎn)GPU企業(yè)布局資本市場(chǎng)加速 輔導(dǎo)工作完成

吳艷妮宣布:捐款20萬,!助力患兒康復(fù)希望

專家預(yù)測(cè)國足2034年進(jìn)世界杯 孫繼海:十年左右

一根頭發(fā)賣了6萬元,?“成功學(xué)大師”詐騙案宣判

美國空襲后,伊朗最高領(lǐng)袖首次發(fā)聲 必須受到懲罰

印度防長來華參會(huì)有何用意 釋放“選邊站”信號(hào)

果然全世界都被騙了,!美軍正式參戰(zhàn),!伊朗真正的危險(xiǎn)來了! 哈梅內(nèi)伊政權(quán)無路可走
今日五月廿八,,“見三空”指得啥,?

諧音梗商戰(zhàn)為何出圈,?原來商戰(zhàn)可以這么不正經(jīng)

NBA總決賽G7:步行者vs雷霆 王牌控衛(wèi)決戰(zhàn)搶七之巔,!背水一戰(zhàn)創(chuàng)歷史

伊朗核設(shè)施受損情況如何 初步評(píng)估顯示破壞但未摧毀

伊朗伊斯法罕離心機(jī)制造車間遇襲 一周內(nèi)第三次襲擊

德國乒協(xié)前總教練點(diǎn)贊樊振東:相信他仍然會(huì)處于頂級(jí)水平

美國增派驅(qū)逐艦赴東地中海 強(qiáng)化以色列防御態(tài)勢(shì)
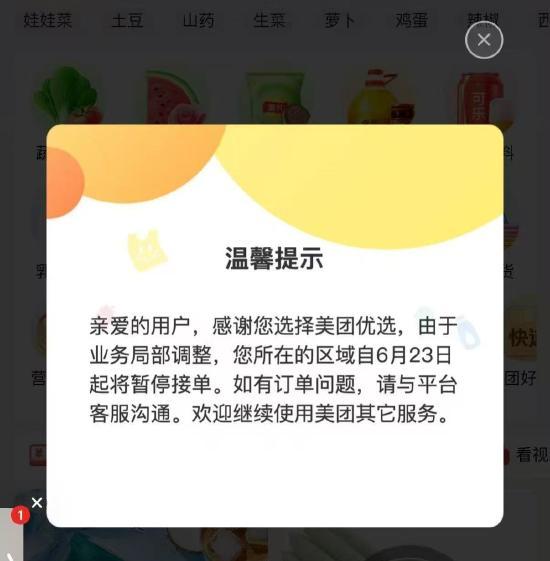
美團(tuán)優(yōu)選關(guān)閉 業(yè)務(wù)調(diào)整聚焦優(yōu)勢(shì)區(qū)域

亂打開了,約旦伊拉克敘利亞都在開火,,槍口不對(duì)準(zhǔn)以色列卻打伊朗 中東國家立場(chǎng)矛盾

特朗普犯下大錯(cuò),!美軍打擊伊朗核設(shè)施,美國再次踏入帝國墳場(chǎng),?局勢(shì)恐將升級(jí)

喝茶也能“凈化”水,?研究發(fā)現(xiàn)泡茶過程或能吸附重金屬
吳艷妮宣布:捐款20萬,!助力患兒康復(fù)希望
專家預(yù)測(cè)國足2034年進(jìn)世界杯 孫繼海:十年左右

美國轟炸伊朗核設(shè)施,,將帶來三大影響 中東局勢(shì)緊張升級(jí)

馬龍張雨菲現(xiàn)身奧林匹克日的活動(dòng)現(xiàn)場(chǎng),稱沒實(shí)現(xiàn)的夢(mèng)想還有很多

美烏克蘭事務(wù)特使罕見訪問白俄羅斯,,美媒:希望“盧卡申科能幫助俄烏達(dá)成可能的和平協(xié)議”,;白宮高級(jí)官員五年多來首訪

胡塞武裝向以色列發(fā)動(dòng)導(dǎo)彈襲擊 局勢(shì)升級(jí)引關(guān)注

聯(lián)合國專家譴責(zé)以色列打擊伊朗 加劇中東動(dòng)蕩不安

媒體評(píng)寧波東方理工大學(xué)學(xué)費(fèi)9.6萬 高額學(xué)費(fèi)引熱議

男子水中遇5米大白鯊,轉(zhuǎn)身游走還是原地停留,?為何鯊魚傷人后從不獵食

美媒記者探訪特拉維夫看到滿目瘡痍毀滅景象

美國參戰(zhàn),!伊以沖突結(jié)局將如何,? 局勢(shì)升級(jí)或促和平談判
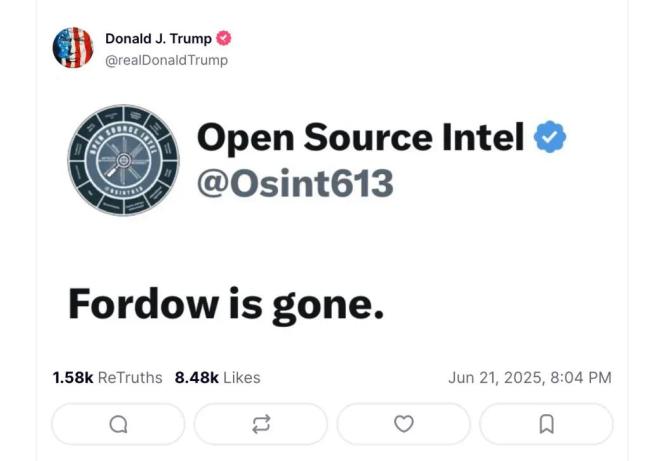
空襲伊朗,美軍欺騙了全世界,! 戰(zhàn)術(shù)欺騙大獲成功

華僑大學(xué)校長寄語畢業(yè)生 敢為人先勇追夢(mèng)
沐曦股份完成IPO輔導(dǎo) 國產(chǎn)GPU企業(yè)布局資本市場(chǎng)加速 輔導(dǎo)工作完成

美空襲伊核設(shè)施 以色列突襲引發(fā)地區(qū)緊張局勢(shì)升級(jí)

美軍轟炸伊朗核設(shè)施,,特朗普發(fā)表全國講話 核設(shè)施被徹底摧毀

男孩查中考成績緊張到撫胸深呼吸,看到分?jǐn)?shù)后笑了“863”,!
相關(guān)新聞
全球首例,!基因編輯豬肝臟,被移植到人體
2025-03-27 14:55:50全球首例,!基因編輯豬肝臟英國發(fā)現(xiàn)綿羊感染禽流感病毒 全球首例引發(fā)關(guān)注
2025-03-26 02:15:37英國發(fā)現(xiàn)綿羊感染禽流感病毒重拳出擊加拿大 中國采取全球首例反歧視措施
2025-03-08 10:19:37重拳出擊加拿大全球首例羊確診禽流感 哺乳動(dòng)物感染引擔(dān)憂
2025-03-30 16:08:54全球首例羊確診禽流感全球首例,!中國醫(yī)生將豬肝植入人體 異種移植重大突破
2025-01-10 08:36:46全球首例中國醫(yī)生將豬肝植入人體全球首例同時(shí)換臉換手男子結(jié)婚 愛情超越一切
2018年,,美國新澤西州男子喬·迪梅奧遭遇了一場(chǎng)嚴(yán)重車禍,導(dǎo)致其全身燒傷面積達(dá)80%,,臉部完全毀容
2025-02-12 20:41:44全球首例同時(shí)換臉換手男子結(jié)婚








